 This morning, the FDA published its 17-page first draft of a working model for its pre-certification program for software as a medical device (SaMD). Though still preliminary (the agency is asking for comments by the end of May), the publication comes with some long-awaited details about how the program might work.
This morning, the FDA published its 17-page first draft of a working model for its pre-certification program for software as a medical device (SaMD). Though still preliminary (the agency is asking for comments by the end of May), the publication comes with some long-awaited details about how the program might work.
The document lays out five "excellence principles" on which companies applying for the program would be evaluated: product quality, patient safety, clinical responsibility, cybersecurity responsibility, and proactive culture.
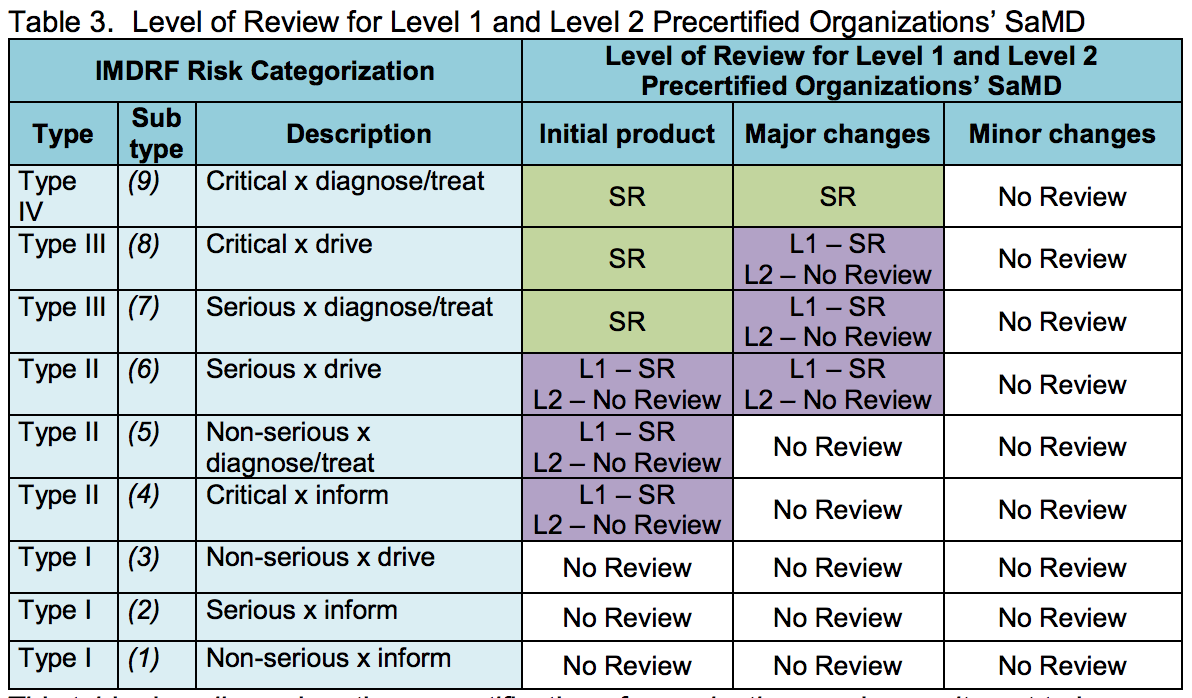
The FDA is proposing two different levels of pre-certification, one for companies with experience putting out SaMD, and one for companies deploying SaMD for the first time. For both categories, once the pre-certification is earned, low-risk devices could be released with no premarket review and higher-risk devices would go through a streamlined process, but exactly where that line is drawn would be different for the two different levels. The risk framework used will be the same framework in the IMDRF guidelines the FDA adopted in December.
"The goal of establishing levels of certification is to allow organizations at different stages in achieving excellence to afford the advantages of the program that is commensurate with their level," the agency wrote.
The document says that once pre-certification is earned, maintenance of Pre-Cert status will be automatable. The framework will also include real world performance data collected by companies and regularly shared and reviewed by the FDA, allowing the agency to keep tabs on devices in the market.
"SaMD manufacturers have the capacity to continuously improve by leveraging knowledge obtained through the ongoing monitoring, collection, and analysis of SaMD product performance," the agency wrote. "To verify the ongoing safety and effectiveness of SaMD products marketed through the Pre-Cert Program, all precertified organizations will be required to demonstrate a robust program for monitoring real world performance data related to their SaMD devices, and for sharing such data with FDA."
In an email to MobiHealthNews, Bradley Merrill Thompson, a partner at Epstein Becker Green who specializes in FDA law, said the document is asking the industry to give the FDA a lot of power and control.
"When someone knocks on your door and says I’m from Washington and I’m here to help you, make sure you read the fine print," he wrote. "In the case of FDA’s newly developed software precertification program, the cost for industry is pretty clear. FDA wants more power. More authority. This is a good old-fashioned trade. Industry wants faster approvals. FDA wants more control over industry. So FDA’s proposing an exchange: faster approvals for more FDA authority."
That manifests itself in several different ways throughout the guidance, Thompson argues. Software that previously fell under enforcement discretion will now be part of the framework. Premarket reviews, while expedited, will delve deeper than they have previously. And the FDA, if the plan goes forward, would have authority to access more of a company's records, not to mention demanding access to companies' real-world data.
"Historically we’ve given FDA much greater power over the premarket decision, and much less power over the postmarket evaluation," he wrote. "If we want to fundamentally change that formula, industry needs to really think about where an FDA person, with the wrong motivation, could cause problems that would ultimately compromise access to care for patients."
There's still opportunity for the industry to give input on this working model until May 30th. The agency will also host an interactive webinar May 10th to discuss the working model and next steps.
"I’m not at this juncture saying that any of FDA’s requested authority to increase its power is necessarily wrong," Thompson said. "But I am saying that industry has to review this proposal with eyes wide open."


















